식약처, 제네릭 경쟁력 강화 민간협의체 운영 결과 공개 함량 달라도 생동성 입증해야…원료 단계부터 현미경 조사
식품의약품안전처가 의약선진국 대비 떨어지는 제네릭(복제약)의 처방 현황 등을 개선하기 위해 21개 과제를 도출했다.
함량이 다른 제품간에도 '비교용출시험'이 아닌 '생물학적동등성시험'으로 동등성을 입증토록 기준을 강화하고, 의사·약사 연수교육 과정에 의약품 허가관리 제도, 생동시험 결과 통계자료 등을 자료로 제공해 처방 신뢰도를 높인다는 계획이다.
과제별 세부시행 방안16일 식약처는 제네릭의약품의 경쟁력 강화를 위해 지난 5월부터 구성·운영해온 '제네릭의약품 국제경쟁력 강화를 위한 민관협의체' 운영에 따라 이같은 내용을 공개했다.
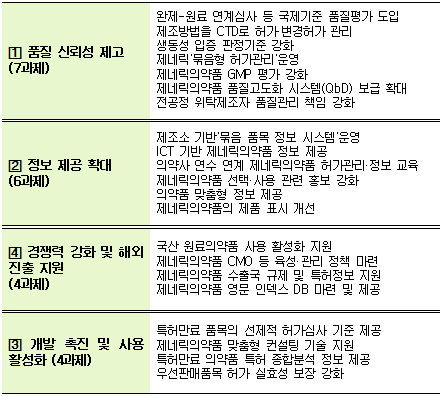
민관협의체에서는 제네릭의약품의 품질, 안전관리 및 국제경쟁력 강화 방안에 대해 집중 논의했으며, ▲제네릭의약품의 품질신뢰성 제고 ▲정보제공 확대 ▲제품 개발 촉진 ▲K-제네릭 해외진출 지원 등 4개 분야 21개 세부과제별 향후 추진 계획을 마련했다.
주요 과제들은 먼저 제네릭의약품 품질신뢰성 제고를 위해 품질이 확보된 제네릭의약품이 유통될 수 있도록, 전 공정을 위탁해 제조하는 때도 GMP 적격성 여부를 사전에 확인하고 허가하도록 했다.
현행 제약사는 자사 품목을 위탁제조품목하는 경우 GMP 자료 면제 대상이었다. 실제 제조하는 수탁자 품목만 GMP 자료를 제출해야 했지만 식약처는 이를 개선, 위탁제조품목도 허가 시 3개 제조번호를 실제 생산 후 GMP 자료를 제출해야 한다.
한편 제네릭의약품의 허가 이후 품질 및 약효에 영향을 미칠 수 있는 세부공정 변경, 원료의 입자크기 변경 등도 업체 자율적 관리에서 정부 주도 관리 체계로 바뀐다.
식약처는 제약사의 '제조방법' 변경 시 품질·약효 영향에 따라 변경 정도를 차등화해 사전 변경허가 하는 체계로 변경했다. 제네릭의약품의 동등성 유지를 위해 주성분과 제형이 동일하고 함량이 다른 의약품의 경우, 기존에는 비교용출시험 자료 제출이 필요했지만 바뀐 기준에서는 함량이 다른 제품 간의 첨가제의 종류·배합비율이 다를 경우 '생물학적동등성시험' 자료를 제출해 동등성을 입증해야 한다.
원료에서 기인한 불순물 혼입 사태에 따라 완제품 이전 원료 단계에 대한 현미경 조사도 병행된다.
식약처는 원료의약품 등록 이후 완제의약품의 품질심사를 진행하던 것을 완제의약품 심사 시 원료의약품에 관한 품질심사를 병행하는 방식으로 변경했다.
이를 통해 완제의약품 심사단계에서 원료의약품 관련 불순물(NDMA 등)과 같은 위험요인을 사전에 파악하는 등 의약품의 품질 사각지대를 해소할 수 있다는 계획이다.
또 국산 원료 사용 제네릭의약품 개발 촉진을 위해 국산 원료의약품 사용 시 완제의약품을 신속히 심사하는 인센티브를 제공할 예정이다.
식약처는 "의약품 허가관리 제도, 생동시험 결과 통계자료 등을 교육자료로 제공해 의약사의 신뢰도 향상을 꾀하겠다"며 "오리지널의약품 특허에 도전하는 업체를 지원함으로써 제네릭의약품 개발을 촉진하는 방안도 함께 추진된다"고 덧붙였다.
댓글은 로그인 후 댓글을 남기실 수 있으며 전체 아이디가 노출되지 않습니다. ex) medi****** 아이디 앞 네자리 표기 이외 * 처리 댓글 삭제기준
다음의 경우 사전 통보없이 삭제하고 아이디 이용정지 또는 영구 가입이 제한될 수 있습니다.
1. 저작권・인격권 등 타인의 권리를 침해하는 경우
2. 상용프로그램의 등록과 게재, 배포를 안내하는 게시물
3. 타인 또는 제3자의 저작권 및 기타 권리를 침해한 내용을 담은 게시물
4. 욕설 및 비방, 음란성 댓글